
Before the introduction of the Common Technical Document (CTD) in 2002, each major regulatory region, including the European Union (EU), the USA, and Japan, had its unique guidelines and formats for submitting regulatory dossiers to obtain marketing approval for new drugs or changes to existing licenses. For instance, Japan required the GAIYO, Europe needed Expert Reports and Tabulated Summaries, and the USA had specific FDA guidelines for New Drug Applications (NDA). These diverse requirements within the EU further complicated submissions, making the process highly repetitive and time-consuming across different countries and regions.
In 2000, to streamline this process, representatives from the European Medicines Agency (EMA), the USA FDA, and Japan’s Ministry of Health, Labour, and Welfare under the International Conference on Harmonisation (ICH) umbrella created unified guidelines for these dossiers. The CTD, first issued in 2002, aimed to reduce the time and resources needed to compile applications, simplify regulatory reviews, and improve communication with applicants through a standard format. It became the mandatory format for NDAs in the EU and Japan by July 2003 and was strongly recommended in the USA. Other countries including Canada and Switzerland have since adopted the CTD and are transitioning to an electronic format, the eCTD, mandatory in the EU for centralized procedures since 2010.
General Principle of CTD

Overall organization of CTD
The CTD’s overall structure, outlined in the ICH M4 guidelines, includes a section detailing document location and pagination within the dossier, known as granularity. This guidance is beneficial for dossiers containing multiple indications or components of the investigational medicinal product (IMP). Additionally, question-and-answer documents accompany the M4 guidelines to address common issues. The CTD dossier comprises five main modules: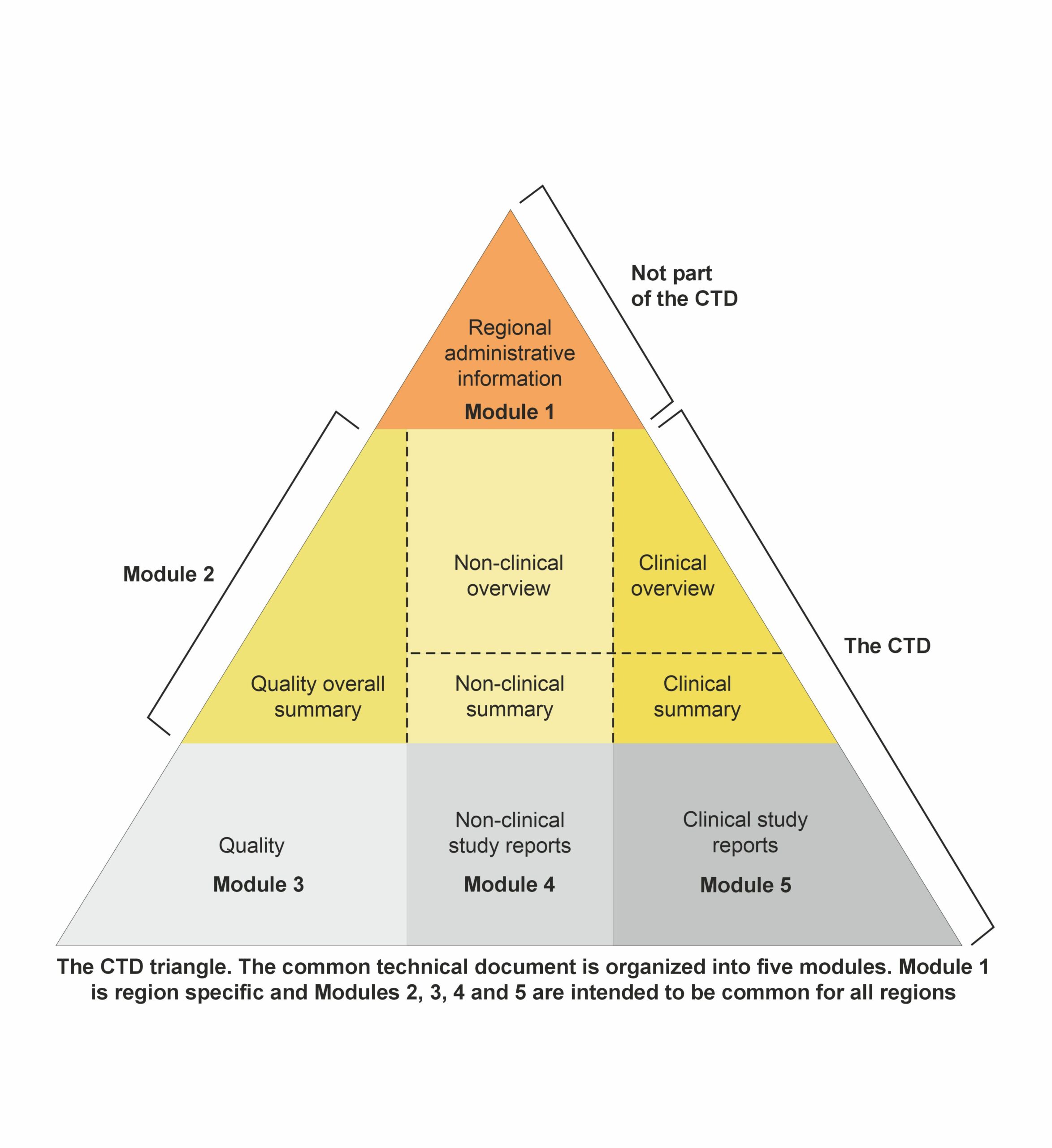 Module 1: Administrative and prescribing information
Module 1: Administrative and prescribing information
It is region-specific and includes all administrative documentation and prescribing information required by the local regulatory authority. This module comprises application forms, the proposed labeling, environmental risk assessments, and manufacturer details tailored to meet local legal and regulatory mandates. It sets the stage for the evaluation process by providing essential administrative and information.
Modules 2–5though, are standard to all regions, and these comprise the main body of the CTD.
Module 2: Overviews and Summaries of Modules 3–5
Module 2 consists of seven sections organized as follows:
Provides a brief overview of the IMP, including its pharmacological class, mode of action, and proposed clinical use. In general, the introduction should not exceed one page.
Click Here:- The Power of Visualization: How Scientific Diagrams, Graphs, Videos, Infographics Boosts Publications and Citations
Module 3: Quality documentation
It contains the chemistry, manufacturing, and controls reports essential for the product’s registration dossier. The specific requirements for Module 3 are outlined in detail in the ICH M4Q guideline. This module comprises sections covering both the drug substance and the drug product. The main headings within Module 3 must remain unchanged.
Module 4: Non-clinical reports on pharmacology and toxicology
Module 4 comprises the non-clinical reports incorporated into the dossier. The organization and content of Module 4 are delineated in the ICH M4S guidelines. The primary headings within this section should remain unchanged.
Module 5: Clinical study reports
Module 5 showcases the clinical reports incorporated into the dossier. The organization and content of Module 5 are outlined in the ICH M4E guidelines, which offer precise instructions for the placement of clinical study reports and related information to streamline preparation and review processes and guarantee completeness. A report’s positioning is dictated by the study’s primary objective, with each report appearing in only one section. In cases of multiple objectives, the study should be cross-referenced in the appropriate sections. The principal headings within this section, which should remain unchanged, include:
This module is pivotal in demonstrating the drug’s therapeutic benefits outweigh its risks, supporting its clinical utility.
Conclusion
The CTD format is a cornerstone of modern drug regulatory submissions, facilitating a smoother, more efficient review process that benefits regulatory authorities, pharmaceutical companies, and ultimately, patients. By standardizing the format and content of drug dossiers, the CTD helps ensure that high standards are maintained, and that new, effective, and safe medicines are made available to the public promptly. Understanding and mastering the structure of the CTD is essential for any pharmaceutical professional involved in the global submission of drugs for approval.
For those interested in taking their first steps into medical writing or enhancing their expertise to meet the challenges of omnichannel communication in healthcare, we invite you to explore the opportunities available through our training programs. Contact us at [email protected] to learn how you can join the ranks of medical writers making a significant impact in healthcare communication. Together, let’s shape a future where accurate, accessible, and actionable health information reaches every corner of the globe, empowering individuals and transforming healthcare outcomes.